
Pompe Disease
Pompe’s Disease or Glycogen Storage Disease II, is a rare, inherited fatal disease affecting infants. It is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body due to an accumulation of glycogen in the lysosome resulting from a deficiency of the lysosomal acid alpha-glucosidase enzyme. It is the only glycogen storage disease with a defect in lysosomal metabolism, and the first glycogen storage disease to be identified, in 1932 by the Dutch pathologist J. C. Pompe. There is no true treatment, but research is underway for Pompe’s and other similar diseases. Myozyme and Lumizyme are two approved treatments for Pompe’s (Sanofi-Genzyme) that are somewhat effective, but not curative. There are no relevant animal models.
Histopathology of Pompe’s Disease at CBI
CBI has experience preparing and evaluating the pathology from patients with Pompe’s Disease. We have recently developed unique histopathologic and ultrastructural methods to evaluate clinical biopsy samples from patients on clinical trials. We offer the following:
Histology Assessments
- Paraffin, plastic and frozen sections of surgical biopsies from treated and untreated patients
- Special histochemical staining
- Proprietary novel method for PAS staining on Epon thin sections to identify intracellular glycogen
- Immunohistochemistry
- Histomorphometric assessment of intracytoplasmic vacuoles
Electron Microscopy
- Ultrastructural sections with standard evaluation of photomicrographs
- Histomorphometric assessment of intracytoplasmic vacuoles, lysomes and glycogen accumulation
Pathology Assessment
- ACVP board certified veterinary pathology experienced in the evaluation of these lesions
- Experienced research pathologist skilled in ultrastructure and histomorphometry
- Evaluation of histologic and ultrastructural specimens
- Histomorphometry and statistical analysis
- Photomicroscopy
- Complete report preparation
↓ Below are some representative histology with thin sections and TEM of GSD specimens that we prepared and evaluated in our laboratorie laboratory. Digital image analysis and histomorphometry were conducted on the TEM samples below. ↓

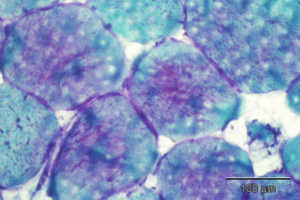

Left: TEM micrograph showing ultrastructural changes in the lysosomes typical of Pompe’s Disease. Middle: PAS on ultrathin plastic sections of a Pompe’s Disease patient. Right: PAS on ultrathin plastic sections of a severely affected Pompe’s Disease patient.
♦
Be sure to check out the movie:
EXTRAORDINARY MEASURES
The film is about parents who form a biotechnology company to develop a drug to save the lives of their children, who have a life-threatening disease. The film is based on the true story of John and Aileen Crowley, whose children have Pompe’s Disease.
Glycogen Storage Disease Type II
Study Details: WHO MAY QUALIFY

Contact Comparative Biosciences, Inc. to discuss a scientific study program to facilitate the evaluation of Pompe Disease Studies and Services.